A detailed process for product registration in Malaysia is available at http://npra.moh.gov.my/ The document below has been compiled specific for natural product or Health supplement only.
A. INTRODUCTION
Under the CDCR 1984, Regulation 7(1): Except as otherwise provided in these Regulations, no person shall manufacture, sell, supply, import, possess or administer any product unless
(a) The product is a registered product; and
(b) The person holds the appropriate licence required and issued under these Regulations.
Under the CDCR 1984, Regulation 2: “Product” means:
(a) a drug in a dosage unit or otherwise, for use wholly or mainly by being administered to one or more human beings or animals for a medicinal purpose; or
(b) a drug to be used as an ingredient of a preparation for a medicinal purpose.
The following criteria may be used as criteria to assist in the classification of products:
- The primary intended purpose of the product
- The primary mode of action/ the principal mechanism of action by which the claimed effect or purpose of the product is achieved;
- Medical device is based on function by physical means, e.g. mechanical action, creation of a physical barrier or replacement or support of organ or body function
- Drug is based on pharmacological, immunological or metabolic action in/on the body
(c) Active ingredients, indication and pharmaceutical dosage form (these are the main criteria for classification of the drugs)
Registration process includes quality control, inspection and licensing as well as post-registration of medicinal products as illustrated below:
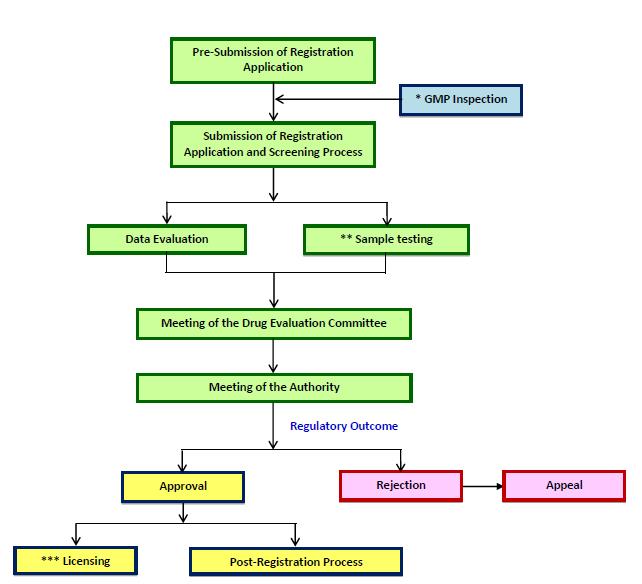
The applicant for product registration shall be known as Product Registration holder (PRH) and must be locally incorporated company, corporate or legal entity with permanent address and registered with Companies Commission in Malaysia (with the scope of business related to the health/pharmaceutical product).
For registration of products, only web-based online submissions via QUEST at https://quest3plus.bpfk.gov.my/front-end/login.php shall be accepted. However, applicant must first register a membership for QUEST system with National Pharmaceutical Control Bureau and purchase a USB Token that contains a User Digital Certificate, from Digicert Sdn. Bhd., which shall be installed to the applicant’s computer. The processing fee and any other charges shall be paid in the form of bank draft/banker’s cheque/money order/postal order made payable to “Biro Pengawalan Farmaseutikal Kebangsaan”.
The process of registration via QUEST 2 will undergo screening process by the Screening Officer. Applicant may need to print the online form as stated in semi manual procedure of screening and submit to:
Ketua Unit Produk Semulajadi,
Pusat Pendaftaran Produk,
Biro Pengawalan Farmaseutikal Kebangsaan,
Lot 36, Jalan Universiti,
46200 Petaling Jaya, Selangor.
Details of the registration process are demonstrated as below:
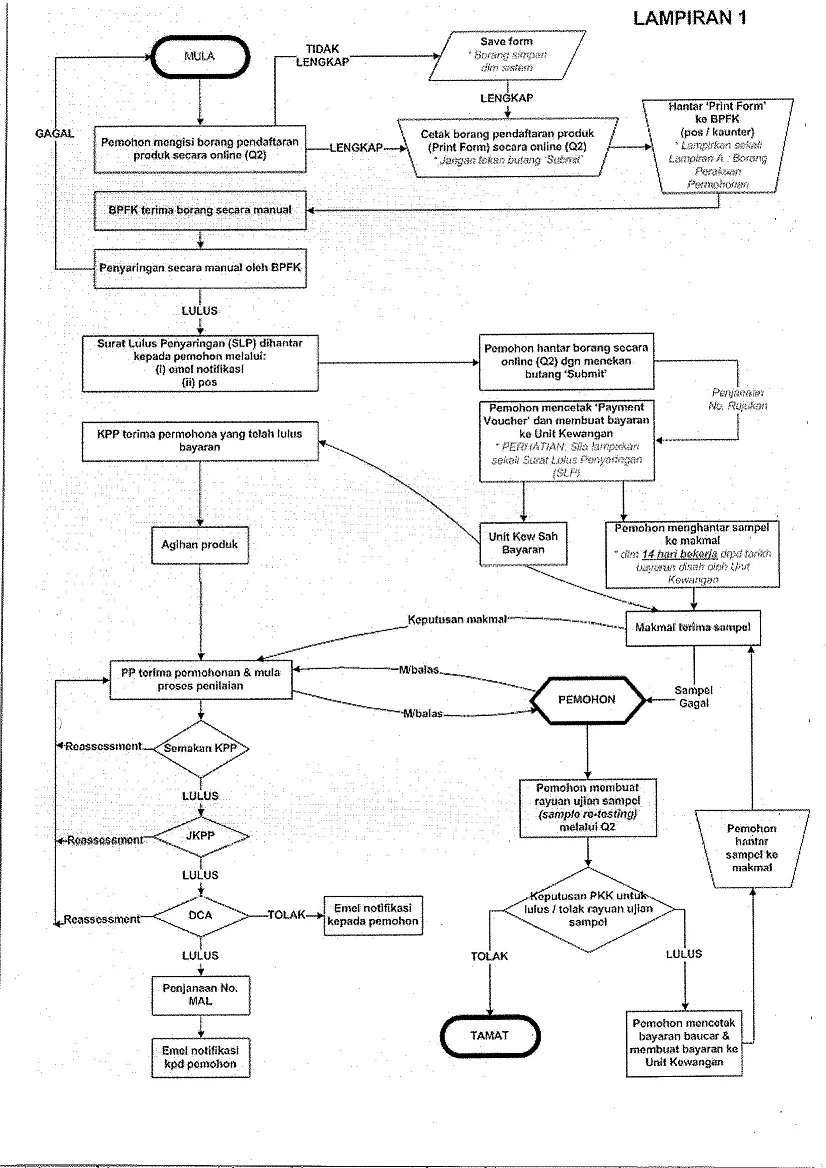
B. PRODUCT REGISTRATION PROCESS- EVALUATION
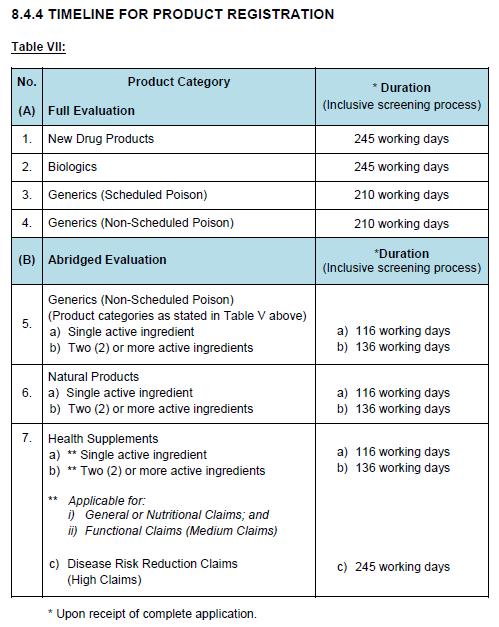
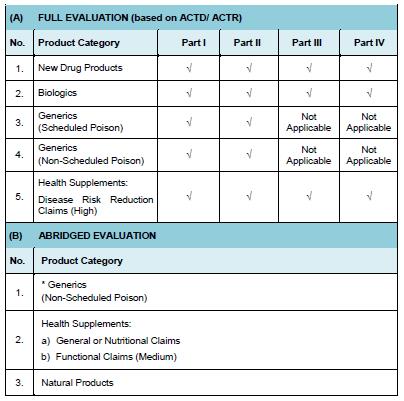
Evaluation of the application is dependently on the product’s category. There are two types of methods applied in the evaluation process that known as full evaluation or abridge evaluation.
Product to be registered under Natural Product or Health supplements with general or nutritional claim or functional claim category must provide dossier consist of elements as below:
GENERAL REQUIREMENTS FOR ABRIDGED EVALUATION
Step 1: Product validation
Step 2:
- Section A: Product particulars
- Section B: Product formula
- Section C: Particulars of packing
- Section D: Label (Mock-up) form immediate container, outer carton, proposed package insert
- Section E: Particulars of product owner, manufacturer, importer and other manufacturer(s) involved and stored address
- Section F: Supplementary documentation
In accordance to ASEAN ACTD/ ACTR or ICH guidelines, categories of product that require a Full Evaluation are:
- New Drug Products
- Biologics Products
- Health Supplements (High claims on disease risk reduction)
GENERAL REQUIREMENTS FOR FULL EVALUATION
Step I: Product Validation
Step II:
- Part I: Administrative Data and Product Information
- Section A: Product Particulars
- Section B: Product Formula
- Section C: Particulars of Packing
- Section D: Label (Mock-up) For Immediate Container, Outer Carton, Proposed Package Insert
- Section E: Supplementary Documentatio
- Part II: Quality of Product
- Section P: Drug Product (Finished Product)
- Section S: Drug Substance
- Part III: Nonclinical Document
- Section A: Table of Contents
- Section B: Nonclinical Overview
- Section C: Nonclinical Written and Tabulated Summaries
- Section D: Nonclinical Study Reports
- Section E: List of Key Literature References
- Part IV: Clinical Document
- Section A: Table of Contents
- Section B: Clinical Overview
- Section C: Clinical Summary
- Section D: Tabular Listing of all Clinical Studies
- Section E: Clinical Study Reports
- Section F: List of Key Literature References, Published Clinical Papers and Latest Periodic Safety Update Report (PSUR)
PART II: QUALITY CONTROL
Documents to be submitted are as below:
Documents to be submitted via online Quest system 1) Complete protocol of analysis (POA) for finished product including preservatives and diluents (if any) 2) Summary of Analytical Method Validation (AMV) include all the relevant validation characteristics, its acceptance criteria and results 3) Certificate of Analysis (COA) for active drug substance (1 batch) and recent batches of finished products (3 batches)
Documents to be submitted as hardcopy: 1) Certificate of analysis for active drug substance (1 batch) and recent batches of finished product (3 different batches) 2) Complete protocol of analysis for active drug substances and finished product (including preservatives and diluents, if any) 3) Complete testing method for the AMV.
Complete results for the AMV with all relevant validation parameters, including acceptance criteria and supporting raw data (e.g. chromatograms, spectrums etc.)
Protocol of analysis (POA)
Details of test methods shall include the following items
i. List of equipment and apparatus; ii. List of chemical, reagents and media; iii.Preparation of solutions such as sample, standard, mobile phase, medium etc. iv.Setting up of analytical instrumentation v. System suitability tests (resolution, percentage of Relative Standard Deviation (%RSD), tailing factor and theoretical plate for High Performance Liquid Chromatography (HPLC) and Gas Chromatography (GC) methods); vi. Complete formula for calculation and interpretation of results; vii. Specification or acceptance criteria
Analytical Method Validation (AMV)
Types of analytical procedures to be validated: a) Identification tests b) Quantitative tests for impurities’ content c) Limit tests for control of impurities d) Quantitative tests of the active ingredient in the sample (assay and dissolution) e) Pyrogen or Bacterial endotoxin test f) Sterility test g) Microbial Contamination Test h) Biological Assay of Antibiotics
Typical validation parameters for chemical tests:
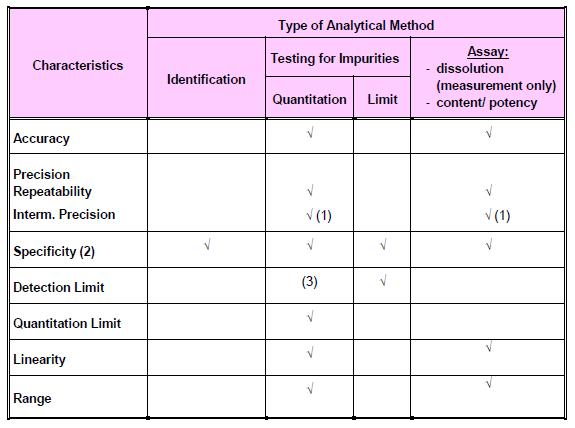
Note: The figure was extracted from Drug Registration Guidance Document (DRGD) available at http://portal.bpfk.gov.my/index.cfm?&menuid=137 access on 24 September 2014.
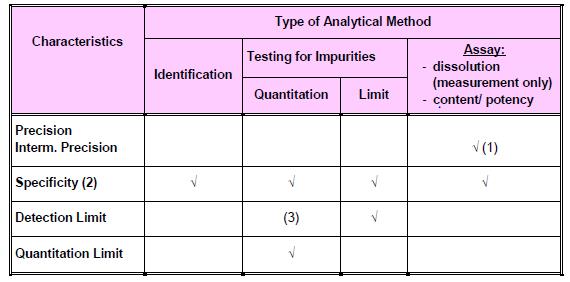
Note: The figure was extracted from Drug Registration Guidance Document (DRGD) available at http://portal.bpfk.gov.my/index.cfm?&menuid=137 access on 24 September 2014.
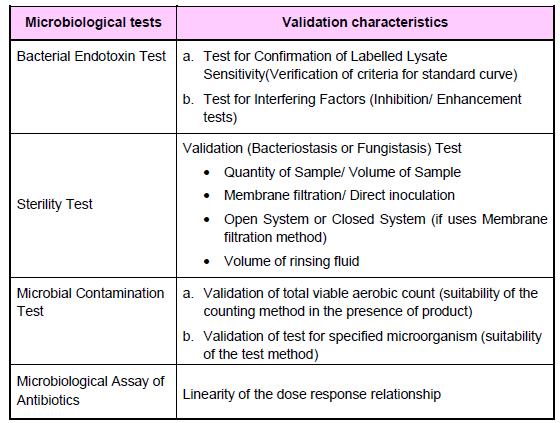
Note: The figure was extracted from Drug Registration Guidance Document (DRGD) available at http://portal.bpfk.gov.my/index.cfm?&menuid=137 access on 24 September 2014.
PART III: NON CLINICAL DOCUMENT
CONTENT OF NON CLINICAL STUDY REPORT:
For ASEAN member countries, the Study Reports of this part may not be required for NCE, Biotechnological Products and other Major Variation Products if the Original Products are already registered and approved for market authorisation in Reference Countries
1) Table of contents
2) Pharmacology
- Primary pharmacodynamics
- Secondary pharmacodynamics
- Safety pharmacology
- Pharmacodynamic drug interactions
3) Pharmacokinetics /ADME
- Analytical methods and validation reports
- Absorption
- Distribution
- Metabolism
- Excretion
- Pharmacokinetic drug interactions
- Other pharmacokinetic studies
4) Toxicology
- Single-dose toxicity (in order by species, by route)
- Repeat-dose toxicity (in order by species, by route, by duration, including supportive toxicokinetics evaluations)
- Genotoxicity
- in vitro
- in vivo (including supportive toxicokinetics evaluations)
- Carcinogenicity (including supportive toxicokinetics evaluations)
- long-term studies (in order by species)
- short- or medium-term studies
- other studies
- Reproductive and developmental toxicity (including range-finding studies and supportive toxicokinetics evaluations)
- Fertility and early embryonic development
- Embryofoetal development
- Prenatal and postnatal development, including maternal function
- Studies in which offspring (juvenile animals) are dosed and/or further evaluated
- Local tolerance
- Other toxicity studies (eg: antigenicity, immunotoxicity, mechanistic studies, dependence, metabolites, impurities)
5) Integrated overview and conclusions (supporting the safety of the product for the intended clinical use)
6) List of Key literature referrences
Non-Clinical Data as below are applicable to disease risk reduction claims:
(For new active ingredient, new combination of active ingredients and new dose)
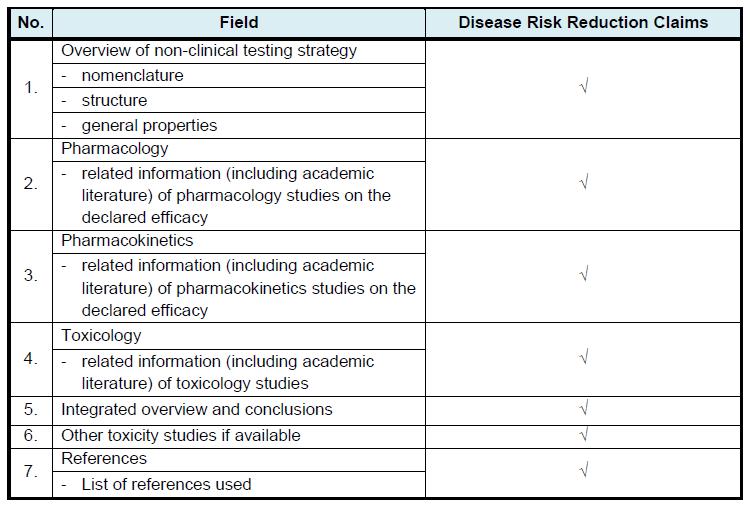
All information must be provided in the following format/ table:
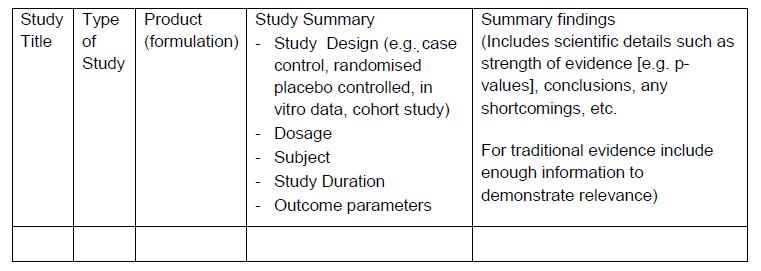
PART IV: CLINICAL DOCUMENT
Clinical Data as below are applicable to disease risk reduction claims:
(For new active ingredient, new combination of active ingredients and new dose).
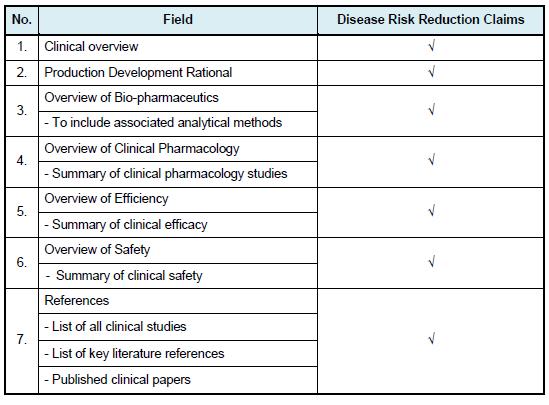
All information must be provided in the following format/table:
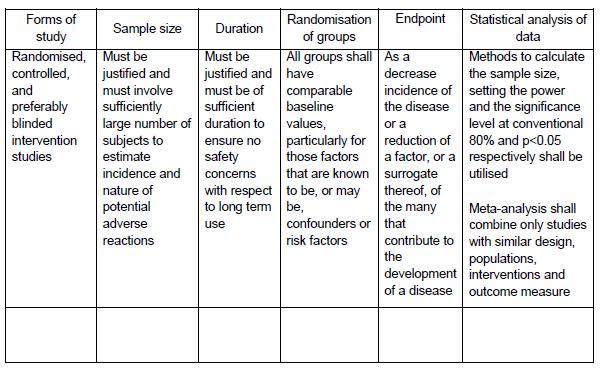
C) REGISTRATION OF NATURAL PRODUCTS
Natural products can be defined as:
a) Traditional medicine (as defined under the Control of Drugs and Cosmetics Regulations 1984): Any product used in the practice of indigenous medicine, in which the drug consist solely of one or more naturally occurring substances of a plant, animal or mineral, of parts thereof, in the unextracted or crude extract form, and a homeopathic medicine. It shall not include any sterile preparation, vaccines, any substance derived human parts, any isolated and characterized chemical substances.
b) Finished Herbal Product Finished herbal products consist of herbal preparations made from one or more herbs. If more than one herb is used, the term “mixture herbal product” can also be used. Finished herbal products and mixture herbal products may contain excipients in addition to the active ingredients. However, finished products or mixture herbal products to which chemically defined active substance have been added, including synthetic compounds and/ isolated constituents from herbal materials, are not considered to be herbal.
c) Herbal Remedy Any drug consisting of a substance or a mixture of substances produced by drying, crushing or comminuting, but without subjecting to any other process, a natural substance or substances of plant, animal or mineral origin, or any part of such substance or substances.
d) Homeopathic Medicine Any pharmaceutical dosage form used in the homeopathic therapeutic system in which diseases are treated by the use of minute amounts as of such substances which are capable of producing in healthy persons symptoms similar to those of the disease being treated.
General requirements for registration of natural products are simplified as below:
D. REGISTRATION OF HEALTH SUPPLEMENTS
A Health Supplement (HS) means any product that is used to supplement a diet and to maintain, enhance and improve the health function of human body. It may contain one or more, or the following combination:
- Vitamins, minerals, amino acids, fatty acids, enzymes, probiotics, and other bioactive substances;
- Substances derived from natural sources, including animal, mineral and botanical materials in the forms of extracts, isolates, concentrates, metabolites;
- Synthetic sources of ingredients mentioned in (i) and (ii) may only be used where the safety of these has been proven.
All claims made for HS shall:
- be consistent with the definition of HS;
- enable consumers to make an informed choice regarding products;
- not be misleading or false;
- support the safe, beneficial and appropriate use of the product;
- maintain the level of scientific evidence which is proportional to the type of claims;
- be for health maintenance and promotion purpose only;
- not be medicinal or therapeutic in nature, such as implied for treatment, cure or prevention of disease.
Types of HS claims are:
- General or Nutritional claims;
- Functional Claims (medium);
- Disease Risk Reduction Claims (high).
For a HS product making a General or Functional Claim on vitamin(s) and/or mineral(s), it must contain minimum of 15% of the Codex Nutrient Reference Value (NRV) per daily dose of the vitamin(s) and/or mineral(s). Other ingredients must be substantiated by the evidences to which it has been supported. For a HS product making Disease Risk Reduction Claim, it must be substantiated by the evidences to which it has been supported.
Note: Always refer Drug Registration Guidance Document (DRGD) for latest substantiated evidences requirements.
Dossier requirements include:
The requirements are simplified as below:
| SECTION A | PRODUCT PARTICULARS |
| SECTION B | PRODUCT FORMULA |
| SECTION C | PARTICULARS OF PACKING |
| SECTION D | LABELLING REQUIREMENTS |
| SECTION E | PARTICULAR OF PRODUCT OWNER, MANUFACTURER, IMPORTER AND OTHER MANUFACTURER |
| SECTION | SUPPLEMENTARY DOCUMENTS |